
Fluorescence spectroscopy is routinely used for studying structural changes in conjugated systems, aromatic molecules, and rigid, planar compounds due to alterations in temperature, pH, ionic strength, solvent, and ligands. A single fluorophore can generate thousands of detectable photons that can be repeatedly excited and detected, making fluorescence spectroscopy is a highly sensitive technique.
- What Is Fluorescence?
- How Does Fluorescence Spectroscopy Work?
- Instrumentation of Fluorescence Spectroscopy
What is Fluorescence?
How Does Fluorescence Spectroscopy Work?
Fluorescence is a type of radiative emission that occurs when a molecule absorbs energy at a wavelength where it has a transition dipole moment. The excitation energy provided to the molecule at the ground state promotes photons to an excited singlet state, where they then decay to the lowest vibrational energy level of this excited singlet state. This energy further relaxes back to the ground state of the molecule, emitting photons in the process, as shown in Figure 1.
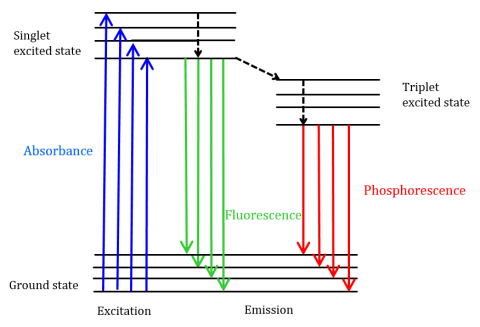
Fluorescent molecules can also undergo there are three methods of nonradiative relaxation where the excitation energy is not converted into photons: (1) internal conversion, (2) external conversion, and (3) intersystem crossing. Internal conversion occurs when there is a relatively small energy gap between two electronic states and the electrons transition from a higher electronic state to one of lower energy. Here the energy is transferred to the vibrational modes of the electronic state. Since vibrational processes are thermally driven, increasing temperature leads to decreases in fluorescence intensity. In external conversion, energy is lost through collisional quenching with solute molecules in the fluorophore’s environment. Intersystem crossing arises when vibrational levels of the singlet and triplet excited states overlap in energy and electrons transition from the lowest singlet excited state to the first excited triplet state. The photons emitted as they return back to the ground state is known as phosphorescence (Figure 1). The triplet state is lower in energy than the singlet state so phosphorescence peaks are found at longer wavelengths than fluorescence. Since these transitions are also forbidden, phosphorescence exhibits a longer lifetime (~10-4 – 102 seconds) compared with fluorescence (~10-9 – 10-6 seconds). The longer lifetimes also lead to thermal deactivation via oxygen quenching, solvent movement, and intermolecular collision so phosphorescence typically cannot be observed at room temperature and samples must therefore be cooled at liquid nitrogen temperature.
Beer’s Law and Concentration Effects
While absorption occurs on the timescale of less than 10-15 seconds, the relaxation process from the excited to the ground state is much slower. Therefore, fluorescence can provide information on a fluorophores’ interactions with surrounding molecules and solvents, unlike absorption.
Fluorescence intensity is directly proportional to the excitation light intensity
F=2.303 * K * I0 * εbc
where K is a constant based on instrument geometry, I0 is the intensity of the excitation light, e is the fluorophore’s molar absorptivity, b is the pathlength, and c is the concentration. Since the fluorescence intensity is not ratioed to the incident light intensity like with absorption measurements, the fluorescence sensitivity is much greater because it is not limited by the instruments ability to differentiate between the incident and detected intensities. Consequently, smaller concentrations are required for measurements.
The above equation is only linear when the sample absorbance is less than 0.05 AU. If a sample is too concentrated, the emission light can be reabsorbed by the fluorophore, attenuating the fluorescence signal at shorter wavelengths. Excitation light may also not fully penetrate the full width of a highly concentrated sample, which will also lead to decreased fluorescence intensities.
Instrumentation of Fluorescence Spectroscopy
Characteristics of a Fluorescence Spectrum
Fluorometers are composed of an excitation and emission monochromator, allowing users to obtain both excitation and emission spectra. A measurement made by a fluorometer is unique to the individual instrument’s excitation and emission monochromators. Fluorescence is directly related to luminous flux and the efficiency of measurement and therefore dependent on the instrument design and components such as the light source, monochromator optics, and photomultiplier tube. Each light source will have a different spectral output (both shape and power) which will vary and decrease over the lifetime of the source.
Excitation spectra plot the intensity at a fixed emission wavelength while varying the excitation wavelengths. Since most emission spectra are independent of the excitation wavelength, the excitation spectra are frequently duplicates of the fluorophore’s absorption spectrum.
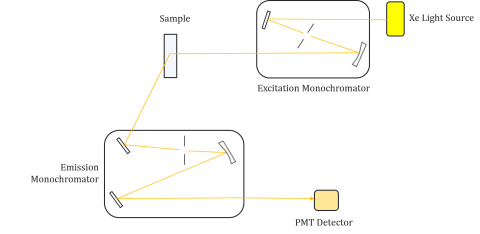
Conversely, an emission spectrum plot the intensity at a fixed excitation wavelength while scanning through varying emission wavelengths. These emission scans provide information on the molecular structure of the fluorophore and the local environment surrounding it. Since the fluorescence emission always occurs from the lowest excited state to the ground state, the shape of the emission spectrum is independent of the excitation wavelength. More energy is also required to excite a molecule from the ground to the excited state, resulting in emission peaks at longer wavelengths (ie smaller energies) than their corresponding excitation wavelengths. This difference in energy between the excitation and emission wavelengths is known as the Stokes shift.
In addition, absorption and emission spectra are frequently mirror images of one another due to the equal distribution between the vibrational energy levels of the excited and ground states (Figure 3). The Franck-Condon principle explains that because the nuclei are relatively large and the electronic transition involved in emission and absorption occur on such fast timescales, there is no time for nuclei to move and the vibrational energy levels and therefore remain roughly the same throughout the electronic transition.
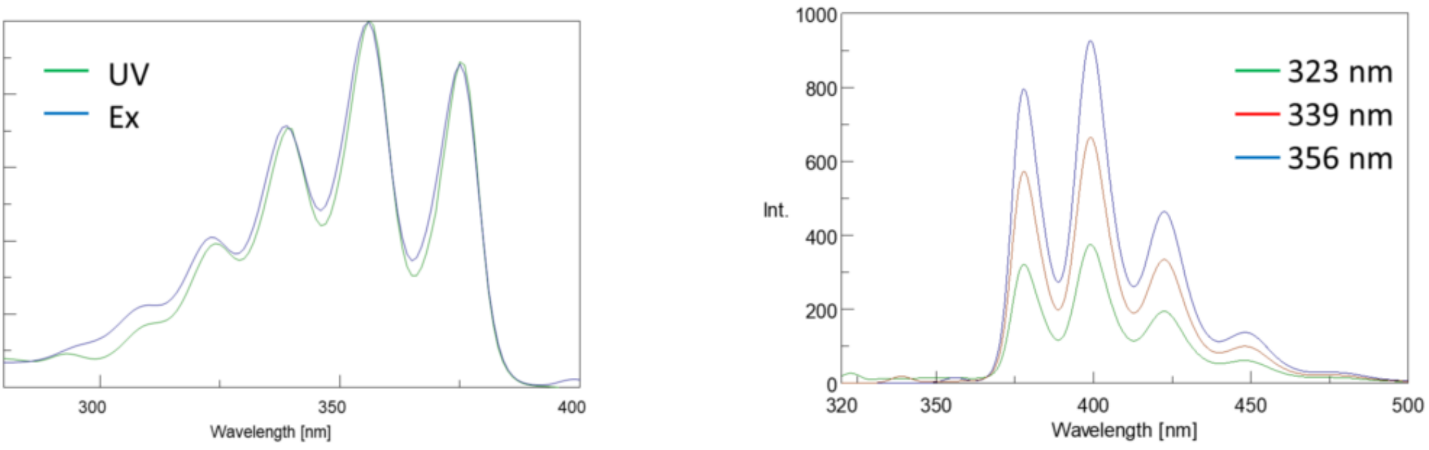
Spectral Bandwidth
Since the fluorescence intensity is proportional to the input light intensity, the amount of light passed through the monochromator will greatly affect the intensity. The sum of the excitation and emission bandwidths should be about the spectral bandwidth (SBW) of the peak being monitored so that all peaks are well resolved. As long as this rule of thumb is followed, the bandwidths can be opened to increase the amount of light throughput for samples with low fluorescence. The SBW can also be impacted by the Stokes shift of the fluorophore. Narrower Stokes shifts may limit the range of acceptable SBWs that can be used.
Fluorescence Artifacts
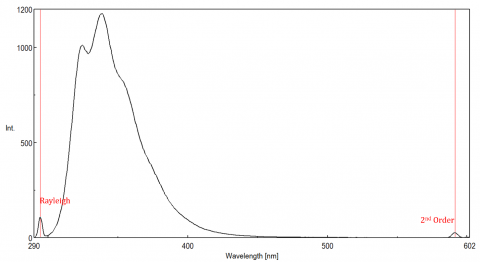
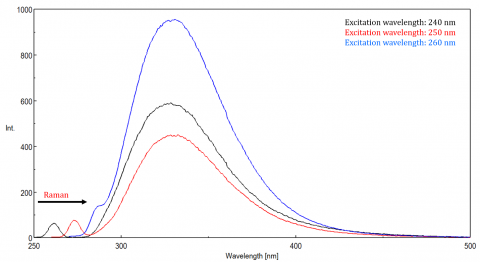
Scattered light can give rise to artifacts, distorting the fluorescence spectrum. The three most common types of scatter seen in fluorescence are Rayleigh, 2nd order, and Raman scatter (Figure 3). Rayleigh scattering is the scattered excitation light and therefore peaks at the excitation wavelength. 2nd order scatter is higher-order scatter observed at twice the excitation wavelength. Raman scattering is inelastic scatter due to solvents and peaks at a fixed energy from the excitation wavelength. To differentiate Raman scattering from a fluorescence peak, the excitation wavelength can be varied in 5 to 10 nm increments and if the peak in question shifts with the excitation wavelength and decreases in intensity, then that peak, is due to Raman scatter. You can also check to see if the peak is in the blank solvent spectrum. If it is, there is a chance it is a Raman peak. If the fluorescence peak is too close or overlapping with either the Raman or Rayleigh scatter, the bandwidths and/or excitation wavelength can be adjusted to shift the scatter off the fluorescence peak. These effects are most prominent for very low fluorophore concentrations and especially highly scattering solutions, like proteins, microsphere, nanoparticles, as well as solids.
Dynamic Range
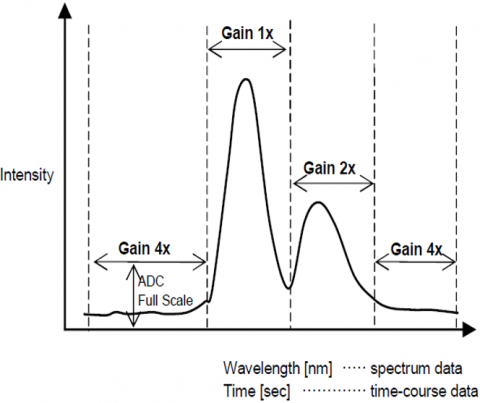
The Automatic Gain Control function automatically adjusts the gain of a signal from the detector based on the fluorescence intensity. This optimizes the signal to noise throughout the entire scanned range for spectral or time course measurements so that peaks with different intensities are automatically adjusted to improve the S/N and assure result accuracy.
Automatic Sensitivity Control System (SCS)
The Automatic Sensitivity Control System(SCS) expands the dynamic range of the detected fluorescence signal by automatically adjusting the detector voltage according to the fluorescence intensity. This allows for fixed wavelength or quantitative analyses measurements of sub-picomolar to micromolar concentrations without manually changing the instrument.
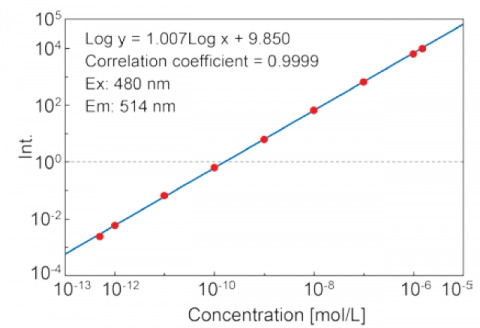
Figure 5. Calibration curve of fluorescein solutions from 5·10-13 to 1.5·10-6 M using the auto-SCS function.